
FineTest
SKU(재고 관리 코드):FNab00330
anti- Alpha galactosidase A antibody
anti- Alpha galactosidase A antibody
Delve into the realm of lysosomal storage disorders with our specialized Anti-Alpha Galactosidase A Antibody, now available in a practical 100µg size. Alpha galactosidase A is a crucial enzyme involved in the breakdown of glycosphingolipids, and its deficiency leads to Fabry disease, a rare genetic disorder. Our meticulously crafted antibody offers exceptional specificity and sensitivity, making it an ideal choice for various applications, from enzyme activity assays to immunohistochemistry.
What truly sets our Anti-Alpha Galactosidase A Antibody apart is its exceptional performance. It has undergone rigorous testing and validation to ensure reliable and reproducible results, effectively reducing experimental variability and saving valuable time and resources.
With a generous 100µg size, this product provides an ideal balance between cost-effectiveness and practicality, offering ample antibody for multiple experiments while minimizing wastage, aligning with our commitment to sustainable research practices.
Engineered for user-friendliness, our Anti-Alpha Galactosidase A Antibody is suitable for researchers of all experience levels, from newcomers to seasoned scientists. It seamlessly integrates into various laboratory techniques, enhancing the precision and reliability of your experiments.
Uncover the mysteries of lysosomal storage disorders with confidence, knowing you have the support of a high-quality, meticulously crafted Anti-Alpha Galactosidase A Antibody. Contribute to groundbreaking insights in the field of rare diseases and enzymology with every experiment.
-
Product Name
Alpha galactosidase A antibody
Size
100µg
Form
liquid
Purification
Protein A+G purification
Purity
≥95% as determined by SDS-PAGE
Host
Mouse
Clonality
monoclonal
Isotype
IgG2a
Storage
PBS with 0.02% sodium azide and 50% glycerol pH 7.3, -20℃ for 12 months(Avoid repeated freeze / thaw cycles.)
BACKGROUND
GLA, also named as Melibiase, Agalsidase and Alpha-galactosidase A, belongs to the glycosyl hydrolase 27 family. It hydrolyzes terminal, non-reducing alpha-D-galactose residues in alpha-D-galactosides, including galactose oligosaccharides, galactomannans and galactolipids. Fabry disease is an X-linked lysosomal storage disorder resulting from the deficient activity of GLA. Enzyme replacement therapy(ERT) with GLA is currently the most effective therapeutic strategy for patients with Fabry disease, a lysosomal storage disease.
IMMUNOGEN INFORMATION
Immunogen
galactosidase, alpha
Synonyms
Alpha galactosidase A, galactosidase, alpha
Observed MW
49 kDa
APPLICATION
Tested Application
ELISA, WB, IHC, IF
Recommended Dilution
WB: 1:500-1:2000; IHC: 1:20-1:200; IF: 1:20-1:200
UNIPROT INFORMATION
UniProt ID
IMAGES
-
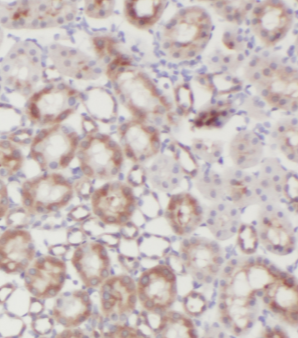 Immunohistochemistry of paraffin-embedded human kidney tissue slide using FNab00330(GLA Antibody) at dilution of 1:50
Immunohistochemistry of paraffin-embedded human kidney tissue slide using FNab00330(GLA Antibody) at dilution of 1:50 -
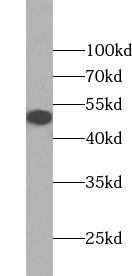 HeLa cells were subjected to SDS PAGE followed by western blot with FNab00330(GLA antibody) at dilution of 1:1000
HeLa cells were subjected to SDS PAGE followed by western blot with FNab00330(GLA antibody) at dilution of 1:1000